

Συμβουλεύουν οι Δημήτριος Χριστόφορος Καραγιάννης, Βιολόγος και Γεώργιος Μαυροθαλασσίτης, Καθηγητής Ιατρικής Χημείας, Ιατρική Σχολή, Πανεπιστήμιο Κρήτης και Συνεργαζόμενος Ερευνητής, Ινστιτούτο Μοριακής Βιολογίας και Βιοτεχνολογίας, Ίδρυμα Έρευνας.
Το σύνδρομο Chitayat (Χιταγιά) αποτελεί μια σπάνια γενετική διαταραχή που συνήθως χαρακτηρίζεται από υπερφαλαγγισμό, δηλαδή ύπαρξη επιπρόσθετων φαλάγγων (οστών) στα δάκτυλα του χεριού εκτός του αντίχειρα, παραμορφώσεις των χαρακτηριστικών του προσώπου κι αναπνευστικές δυσκολίες1. Η αρχική καταγραφή του συνδρόμου πραγματοποιήθηκε από τον Δρ. David Chitayat το 1993, όταν ο ίδιος σε συνεργασία με την ομάδα του αναγνώρισαν την εμφάνιση των συγκεκριμένων συμπτωμάτων σε ένα νεογέννητο αγόρι2. Έκτοτε, 14 ασθενείς έχουν καταγραφεί στη βιβλιογραφία, παρέχοντας περισσότερες πληροφορίες για τα γενετικά αίτια, τον τρόπο κληρονόμησης, αλλά και το φάσμα των συμπτωμάτων του εν λόγω συνδρόμου3-6.
Γενετικά αίτια
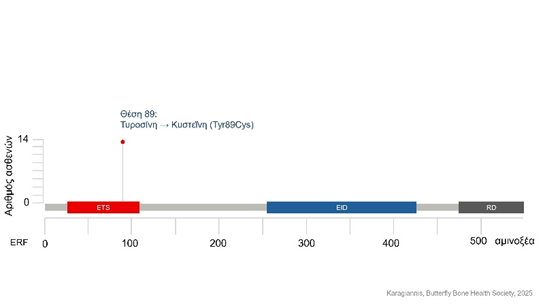
Έως τώρα φαίνεται πως το σύνδρομο Chitayat (CHYTS) προκαλείται από μια συγκεκριμένη μεταλλαγή στο γονιδίωμα του ατόμου, η οποία είναι ίδια για όλους τους ασθενείς κι εντοπίζεται στο γονίδιο ERF (ETS2 repressor factor)3-6. Το γονίδιο αυτό βρίσκεται στο χρωμόσωμα 19 κι είναι υπεύθυνο για την παραγωγή μιας πρωτεΐνης, ονόματι ERF, η οποία κατέχει σημαντικό ρόλο στη γονιδιακή έκφραση, επηρεάζοντας τον κυτταρικό πολλαπλασιασμό, αλλά κι ορισμένα κρίσιμα στάδια κατά την εμβρυογένεση7-12. Η μεταλλαγή, υπεύθυνη για το σύνδρομο Chitayat, αφορά στην αντικατάσταση μιας βάσης αδενίνης (Α) από γουανίνη (G) στο DNA, οδηγώντας στην παραγωγή πρωτεΐνης η οποία στη θέση 89 εμφανίζει το αμινοξύ κυστεΐνη αντί για τυροσίνη3-6. Το αμινοξύ που αντικαθίσταται ως αποτέλεσμα της μεταλλαγής βρίσκεται στην περιοχή πρόσδεσης του ERF με το DNA. Κατ’ αυτόν τον τρόπο, θεωρείται πως δύναται να επηρεάσει την ειδικότητα πρόσδεσης του ERF στο γονιδίωμα, μεταβάλλοντας το ρεπερτόριο επίδρασης του ERF στη γονιδιακή έκφραση.
Η συγκεκριμένη μεταλλαγή εμφανίζεται σε ετερόζυγη κατάσταση, δηλαδή σε ένα από τα δύο αντίγραφα του γονιδίου, και μπορεί είτε να συνιστά αποτέλεσμα κληρονόμησης από γονείς σε παιδιά είτε να παρουσιαστεί χωρίς προηγούμενο ιστορικό στην οικογένεια (de novo μεταλλαγή). Ο τρόπος κληρονόμησης της χαρακτηρίζεται ως αυτοσωμικός κι επικρατής, καθώς ανιχνεύεται σε αυτοσωμικό χρωμόσωμα (οποιοδήποτε χρωμόσωμα εκτός των φυλετικών Χ και Y) κι επικρατής, δεδομένου ότι μεταλλαγή σε ένα από τα δύο αντίγραφα του ERF αρκεί για την εκδήλωση των συμπτωμάτων3-6.
Η πρωτεΐνη ERF αποτελείται από 3 διακριτές περιοχές.
- ETS: χρησιμεύει στην πρόσδεση του ERF στο DNA
- EID: ενισχύει την αλληλεπίδραση του ERF με άλλες πρωτεΐνες
- RD: είναι απαραίτητη για τον ρόλο του ERF στη ρύθμιση της έκφρασης άλλων γονιδίων
Στατιστικά
Λόγω του ότι η μεταλλαγή που ευθύνεται για το σύνδρομο Chitayat εντοπίζεται σε αυτοσωμικό χρωμόσωμα και δεν έχει σχετιστεί με τα φυλετικά, η ασθένεια μπορεί να εμφανιστεί σε ανάλογο ποσοστό σε άνδρες και γυναίκες. Αν κι ο αριθμός των ασθενών που έχουν ανιχνευθεί έως τώρα είναι αμελητέος για την εξαγωγή στατιστικά σημαντικών συμπερασμάτων για τη συχνότητα εμφάνισης του συνδρόμου, αξίζει να αναφερθεί πως η αναλογία ανδρών-γυναικών είναι 1:1.
Συμπτώματα
Το σύνολο των συμπτωμάτων κι η σοβαρότητα τους μπορεί να διαφέρουν ελαφρώς μεταξύ των ασθενών. Παρακάτω παρατίθενται τα πιο χαρακτηριστικά συμπτώματα για το σύνδρομο Chitayat.
Σκελετικές δυσπλασίες
- Υπερφαλαγγισμός (polyphalangy): τα δάκτυλα που μπορεί να επηρεαστούν είναι τα II-V, αλλά συνήθως εμφανίζεται στα II και III13. Λόγω της άτυπης ανάπτυξης της φάλαγγας στην μετακαρποφαλαγγική άρθρωση του δείκτη, πολλές φορές συνοδεύεται με κάμψη του δακτύλου (ulnar deviation), ενώ μπορεί να παρουσιάζεται και σε συνδυασμό με υποπλασία της μεσαίας φάλαγγας (βραχυδακτυλία)14.
- Κλινοδακτυλία: κάμψη του δακτύλου προς τα μέσα, εμφανίζεται κυρίως στο μικρό δάκτυλο του χεριού κι οφείλεται σε άτυπη ανάπτυξη της μεσαίας φάλαγγας
- Κότσι (hallux valgus): διόγκωση της άρθρωσης στη βάση του μεγάλου δακτύλου του ποδιού, συνήθως συνοδεύεται από ερυθρότητα του δέρματος και πόνο
- Σκαφοειδής θώρακας (pectus excavatum): μη φυσιολογικό σχήμα του στέρνου, εμφάνιση βύθισης
Παραμορφώσεις των χαρακτηριστικών του προσώπου
- Εξόφθαλμος: προεξοχή των οφθαλμών από την εσοχή του κρανίου
- Υπερτελορισμός οφθαλμών: αυξημένη απόσταση μεταξύ των κόγχων των ματιών
- Πλατιά κι επίπεδη ρινική ράχη (άνω μέρος της μύτης)
- Φαρδιά χείλη
Οι δύο τελευταίες παραμορφώσεις συνιστούν φυσιολογικά χαρακτηριστικά κι αποτελούν ενδείξεις για ορισμένες γενετικές και κληρονομικές παθήσεις μόνο σε συνδυασμό με άλλα συμπτώματα.
Αναπνευστικές επιπλοκές
- Βρογχομαλακία: κατάρρευση τοιχωμάτων των βρόγχων λόγω υποπλασίας ή απουσίας χόνδρου, εμφανίζεται κυρίως κατά τη βρεφική ηλικία15
Αναπτυξιακές δυσκολίες
- Καθυστέρηση στην ομιλία
- Δυσκολίες λεπτής (π.χ. γράψιμο, σύλληψη αντικειμένων) κι αδρής (π.χ. συντονισμός, ισορροπία) κινητικότητας
Διάγνωση
Τα συμπτώματα του συνδρόμου γίνονται συνήθως αντιληπτά κατά τη βρεφική και παιδική ηλικία. Η διάγνωση μπορεί να πραγματοποιηθεί από ρευματολόγους ή ορθοπαιδικούς χειρουργούς και βασίζεται τόσο στην κλινική εικόνα του ατόμου, όσο και στο ιατρικό ιστορικό της οικογένειας του. Η διάγνωση μπορεί να περιλαμβάνει:
- Ακτινογραφία για τη διαπίστωση ύπαρξης σκελετικών δυσπλασιών
- Βρογχοσκόπηση κι αξονική τομογραφία (CT scan) για διάγνωση βρογχομαλακίας
- Αλληλούχηση γονιδιώματος για την ανίχνευση της μεταλλαγής
Αντιμετώπιση/Θεραπεία
- Χειρουργική παρέμβαση για διόρθωση δυσπλασιών των άκρων
- Υποστήριξη με μηχανικό αερισμό σε περιπτώσεις αναπνευστικής δυσχέρειας
- Λογοθεραπεία για ενίσχυση της γλωσσικής ανάπτυξης
- Φυσικοθεραπεία κι εργοθεραπεία για βελτίωση κινητικών δεξιοτήτων
Πηγές
1. Shoul MI, Ritvo M. Roentgenologic and clinical aspects of hyperphalangism (polyphalangism) and brachydactylism; hereditary abnormal segmentation of the hand. N Engl J Med 1953;248(7):274-8.
2. Chitayat D, Haj-Chahine S, Stalker HJ, Azouz EM, Côté A, Halal F. Hyperphalangism, facial anomalies, hallux valgus, and bronchomalacia: a new syndrome? Am J Med Genet 1993;45(1):1-4.
3. Balasubramanian M, Lord H, Levesque S, Guturu H, Thuriot F, Sillon G, Wenger AM, Sureka DL, Lester T, Johnson DS and others. Chitayat syndrome: hyperphalangism, characteristic facies, hallux valgus and bronchomalacia results from a recurrent c.266A>G p.(Tyr89Cys) variant in the ERF gene. J Med Genet 2017;54(3):157-165.
4. Caro-Contreras A, Alcántara-Ortigoza MA, Ahumada-Pérez JF, González-Del Angel A. Molecular analysis provides further evidence that Chitayat syndrome is caused by the recurrent p.(Tyr89Cys) pathogenic variant in the ERF gene. Am J Med Genet A 2019;179(1):118-122.
5. Shin SH, StJoseph E, Mannan K, Khan K. Radiography of Chitayat syndrome in an infant male. Radiol Case Rep 2019;14(4):448-451.
6. Suter AA, Santos-Simarro F, Toerring PM, Abad Perez A, Ramos-Mejia R, Heath KE, Huckstadt V, Parrón-Pajares M, Mensah MA, Hülsemann W and others. Variable pulmonary manifestations in Chitayat syndrome: Six additional affected individuals. Am J Med Genet A 2020;182(9):2068-2076.
7. Liu D, Pavlopoulos E, Modi W, Moschonas N, Mavrothalassitis G. ERF: genomic organization, chromosomal localization and promoter analysis of the human and mouse genes. Oncogene 1997;14(12):1445-51.
8. de Castro CM, Rabe SM, Langdon SD, Fleenor DE, Slentz-Kesler K, Ahmed MN, Qumsiyeh MB, Kaufman RE. Genomic structure and chromosomal localization of the novel ETS factor, PE-2 (ERF). Genomics 1997;42(2):227-35.
9. Le Gallic L, Sgouras D, Beal G, Jr., Mavrothalassitis G. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol Cell Biol 1999;19(6):4121-33.
10. Le Gallic L, Virgilio L, Cohen P, Biteau B, Mavrothalassitis G. ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression. Mol Cell Biol 2004;24(3):1206-18.
11. Sgouras DN, Athanasiou MA, Beal GJ, Jr., Fisher RJ, Blair DG, Mavrothalassitis GJ. ERF: an ETS domain protein with strong transcriptional repressor activity, can suppress ets-associated tumorigenesis and is regulated by phosphorylation during cell cycle and mitogenic stimulation. Embo j 1995;14(19):4781-93.
12. Papadaki C, Alexiou M, Cecena G, Verykokakis M, Bilitou A, Cross JC, Oshima RG, Mavrothalassitis G. Transcriptional repressor erf determines extraembryonic ectoderm differentiation. Molecular and cellular biology 2007;27(14):5201-5213.
13. Wood VE. Different manifestations of hyperphalangism. J Hand Surg Am 1988;13(6):883-7.
14. Temtamy SA, Aglan MS. Brachydactyly. Orphanet journal of rare diseases 2008;3:1-16.
15. Finder JD. Primary bronchomalacia in infants and children. The Journal of pediatrics 1997;130(1):59-66.
Συμβουλεύουν οι Δημήτριος Χριστόφορος Καραγιάννης, Βιολόγος και Γεώργιος Μαυροθαλασσίτης, Καθηγητής Ιατρικής Χημείας, Ιατρική Σχολή, Πανεπιστήμιο Κρήτης και Συνεργαζόμενος Ερευνητής, Ινστιτούτο Μοριακής Βιολογίας και Βιοτεχνολογίας, Ίδρυμα Έρευνας.



{kind=link}